This blog is a record of my findings and results as an Undergraduate Research Assistant in the Computer Science department of The College of New Jersey from 2014-2015. Much thanks to the school, department, CRA-W, CDC and the NSF for making this possible and thank you for visiting!
|
Because my program only kept both local and global changes that working towards the desired objective (maximization or minimization), I was instructed to modify it so that it only kept the local pieces with a more desirable ∆G and not to make decisions based on the global ∆G. Unfortunately, this modification worsened the extent to which my program optimizes a sequence. For instance, using the GFP protein coding region (whose ∆G = -155.1), my original program maximizes the free energy to -121.4, wheres the modified program only improves it to -149.2. The results are visually represented by the graphs below: ORIGINAL PROGRAM 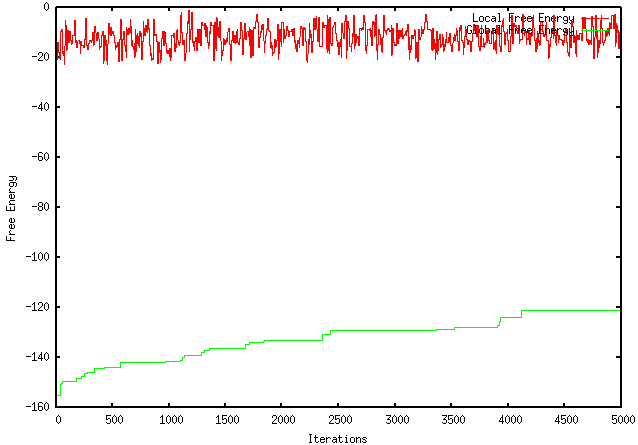 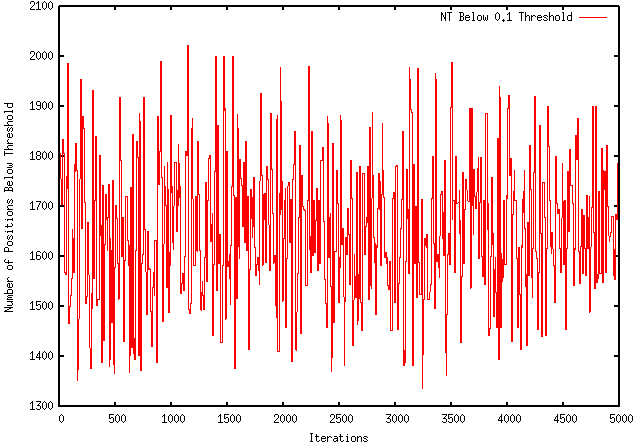 MODIFIED PROGRAM 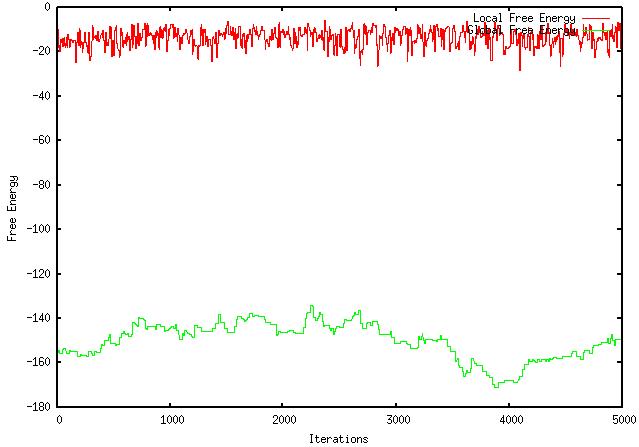 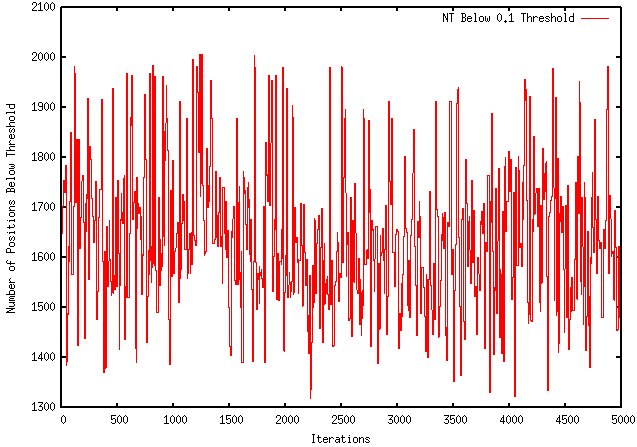 In addition, when using mRNA Optimiser to optimize GFP, it generates a sequence with a ∆G = -106.8. However, it should be noted, when using mRNA Optimiser to optimize GFP while trying to retain GC-content, it produces a sequence with a ∆G = -126.1. Although my program does not try to retain GC-content, it does have an improved ∆G.
This prompted me to examine mRNA Optimiser's method to get an idea about how they go about maximizing the energy more efficiently. First, a number of codons are selected (in every iteration) and changed for synonymous ones. The ∆G of the new sequence is then calculated and "new sequences with larger values are accepted for the next iteration" (similar to my program). However, there is a significant difference, sequences with lower values are accepted (according to the Boltzmann Distribution) to avoid local maxima. According to the paper, it "allows acceptance of worse solutions at the beginning of the process, slowly decreasing the acceptance probability until only better solutions are chosen as the algorithm approaches the maximum number of iterations". The program ends when 4000 iterations are performed. To improve the time in which the program runs, a better method for determining MFE was developed (although it produces less accurate results). RNAFold and MFold/UNAFold both have a time complexity of O(n^3) so mRNA Optimiser developed a more simplistic method and improved it to quadratic complexity. The program itself considers all possible single-stem loop conformations and averages their interaction energy. In addition, there is a detailed method to fine-tuning nucleotide interactions. Moreover, two linear regressions are performed to better predict the MFE. The paper can be found at: http://www.ncbi.nlm.nih.gov/pubmed/23325845. After rerunning my program on the same input sequence for 4000 iterations, my program found a more optimal sequence with a ∆G = -22.5. The sequence is as follows: GTAGTAATTAGATCTGAAAACTTCTCGAACAATGCTAAAACCATAATAGTA CAGCTAAATAAATCTGTAGAAATTAATTGTACAAGACCCAACAATAATACA CGACGCTCAATACATTTTGGACCAGGGAAAGCTTTTTATGCAGGAGAAA TAATAGGAGATATAAGACAAGCATATTGTACACTAAATGGAACTGAATGG AATAATACCTTAAAACAAGTCGCCGAAAAACTAAGAGAACAATTTATAAA AACCATCGTCTTTAATCAATCC I've also generated graphs to visually represent the data collected while my program was running, shown below. To verify that the sequence continued to code for the same protein when translated, http://web.expasy.org/translate/ was used. 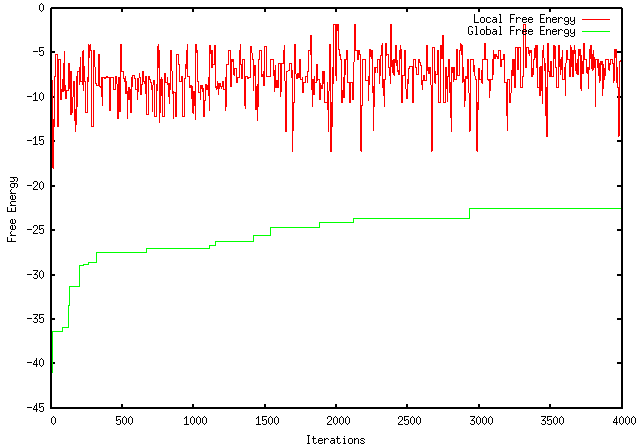 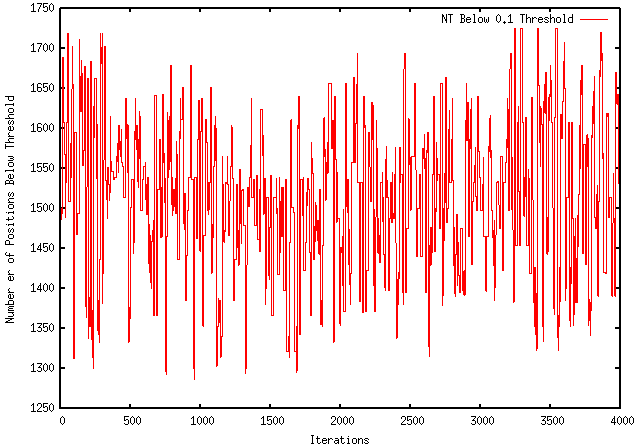 As the end of the semester approaches, I will summarize the program I developed that has generated all of these results.
First, it instantiates 19 variables and opens 3 output files: one for the number of nucleotides above a given threshold (per iteration), one for the local energy (per iteration), and one for the global energy (per 10 iterations). Next it calls importGlobalSequence() which prompts the user for a (FASTA) filename and imports the sequence as a string. Then it passes the global sequence to another subroutine (calculateFoldingEnergy(STRING)) that calculates the folding energy using UNAFold. Next it enters a loop that imports the local sequence (importLocalSequence()) which prompts the user for a window size and creates an array of possible pieces in the sequence and uses "rand" to select a random window. It passes this local sequence to generateFASTAFile() to generate a FASTA file for this sequence and calculates the folding energy of the local sequence using calcFoldingEnergy() again. Then it passes the local FASTA sequence and the name of the desired fold output file and calls Rfold to calculates the local base pairing probabilities. Next is calls createArrOfPosAboveThreshold() which creates an array of positions above (for minimization) or below (for maximization) a user-specified threshold. It then enters another loop (that iterates 10 times) that modify a nucleotide by calling modifyOneNucleotide() which makes a single synonymous changes in positions above/below a threshold by calling synonymousChange(). Then it generates a FASTA file in order to calculate the folding energy and generates an Rfold file to determine the nucleotides above the threshold. the loop repeats, making synonymous changes in positions above/below a given threshold. After this loop, it writes the local energies and number of nucleotides above/below the threshold to 2 output files. Finally, it puts this modified piece back into the global sequence and keeps the changes if it works towards the desired goal (maximizing/minimizing the sequence) and writes the global energy to a third output file. This repeats for however many iterations are desired and outputs the final optimized sequence at the end. After 4000 iterations of mRNA Optimiser on my input sequence, the maximized output sequence generated a maximum folding energy of -23.865 using Mfold. After 4000 iterations of my program on the same input sequence, the maximized output sequence generated a maximum folding energy of -22.7 (also using Mfold). The results and sequences can be summarized in the diagram below: 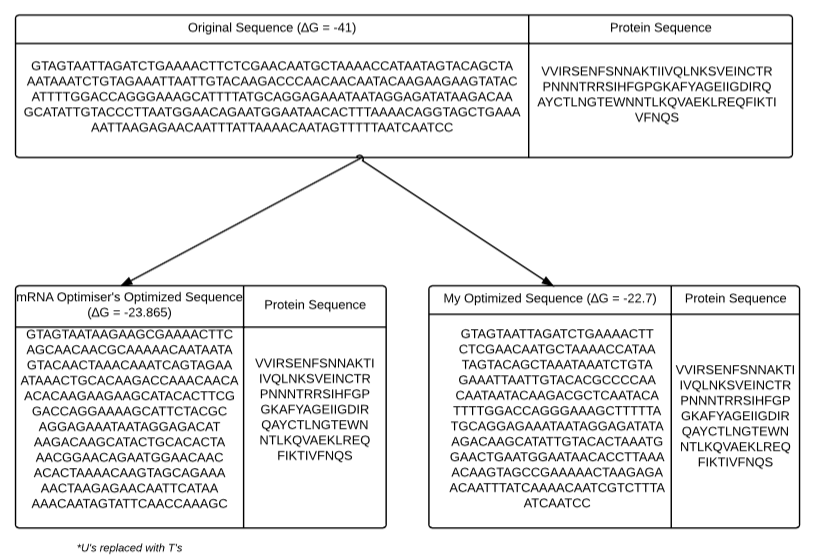 Although my program takes more time than mRNA Optimiser, it can generate synonymous maximized sequences with higher folding energies. It can also minimize the folding energy and allows the user to specify the size of the selected window and Rfold threshold.
For nearly this entire week I have been struggling to understand how to properly use D-Tailor's minimum free energy prediction software. After finally getting it work it, it predicted that the MFE is -2.2, which is very frustrating because it is significantly different than the results from mRNA Optimiser (-8256.7) and Mfold (-41).  I've also began writing up an abstract documenting my results to submit to the ISBRA Conference. However, I need to find other results to compare my results to complete it.
UPDATE: I had access to a PC this evening and was able to install and run Visual Gene Developer. The optimized sequence had a dG = -63.6. These results make the sense in relation to my program's result and provide the most sound comparison. This week I have finished the 1 million iteration run (shown below) and have been working to install and run a program with similar functions to mine so that I can examine my results. 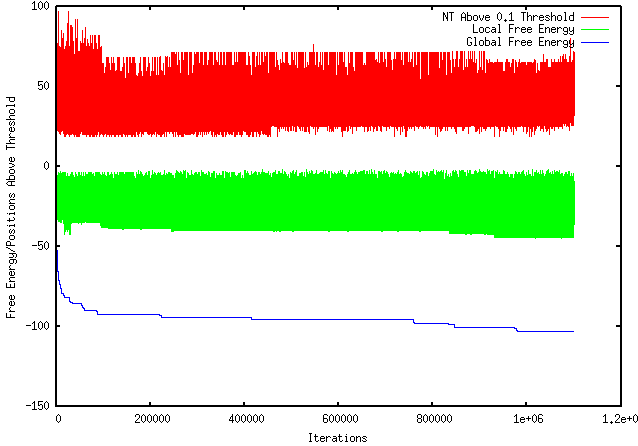 I also read a paper called "Designing RNA Secondary Structure in Coding Regions" by Rukhsana Yeasmin and Steven Skiena that is about optimizing the maximum or minimum folding energies based on local modification. This research is very similar to mine, however, their program is not available for testing. I downloaded and installed a program called D-Tailor to compare my results but am still working on learning Python so that I can design a script that can run it, as you can not run it simply after downloading it.
This week I have run my experiment 4000, 10000, and 1000000000 times. The results are shown below (aside from the 1000000000 iterations which is still running). 4000 Iterations 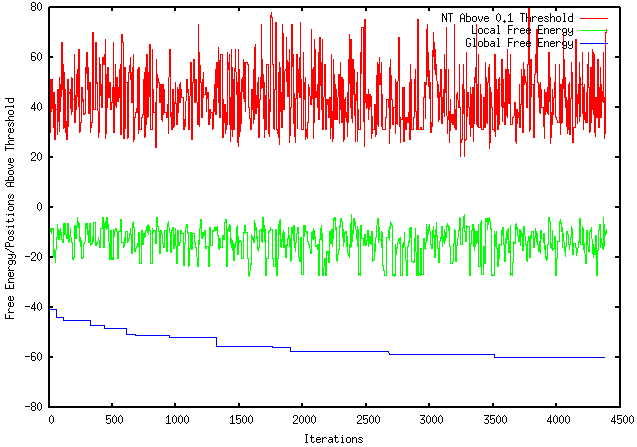 10000 Iterations 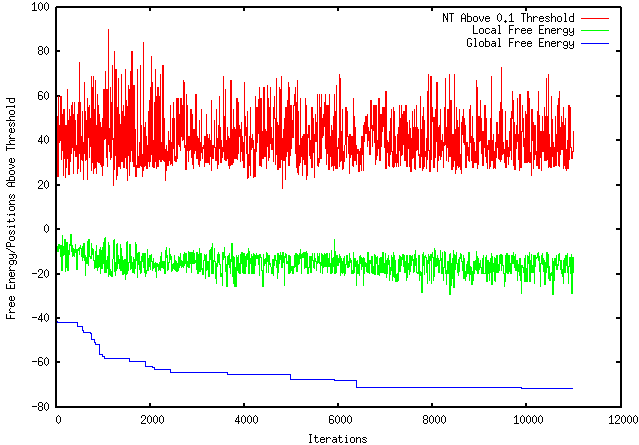 I have compared my results with mRNA Optimiser which continues to provide inconsistent results in comparison to Mfold. mRNA Optimiser ∆G for a sequence:  Mfold ∆G for the same sequence:  After a great, albeit short, spring break, I have continued with my research. This week, I have furthered my research by performing 5 times the amount of iterations as previously performed and comparing my results to an existing mRNA Optimiser. My results are shown in the graph below. 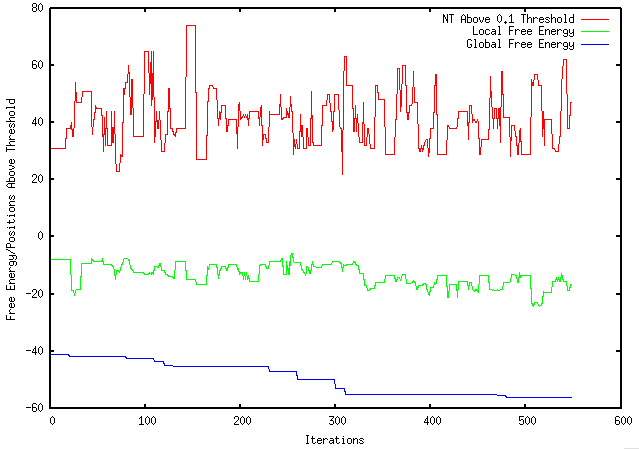 Using a bioinformatics tool called, "mRNA Optimiser", which optimizes mRNA secondary structure, I entered my sequence (after changing Thymine to Uracil) to compare results. After altering the setting to retain the GC content, it found that the initial pseudo-energy (after 4000 iterations) was -8256.7 and the final pseudo-energy was -7076.67. Currently, I am trying to interpret this results as I have found (using Mfold) that the initial folding energy of my sequence is -41 and the optimized energy (after 500 iterations) is -56.
This week I performed more extensive testing and generated results using a sample FASTA file with a DNA sequence as input and my program as output. The results, shown below, are using an Rfold threshold of 0.1 and a window size of 100 nucleotides. 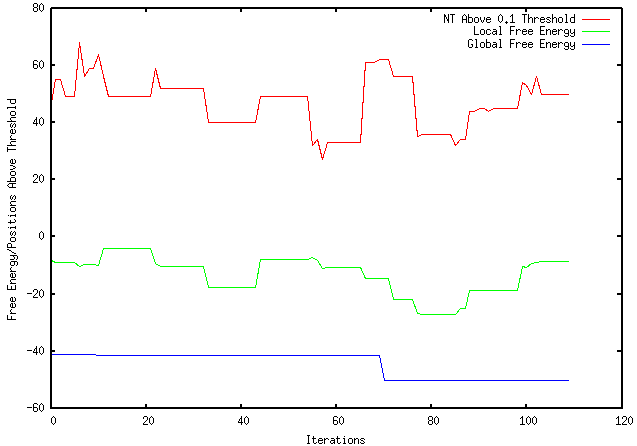 As shown, the top red line represent the number of nucleotides above the 0.1 threshold for each iteration. The middle green line represents the individual local folding energy for that 100-nucleotide sequence. The bottom blue line represents the global energy once the optimized local sequence was put back into the global sequence, which occurred after every 10 iterations. It should be noted that only the lowest global energy was kept, therefore, if there was an instance where the optimized local sequence increased the overall global energy, the lowest global energy from the previous iterations were kept.
I also learned many more features that GNUPlot offers. As you can see, I learned how to plot multiple lines on one graph, manipulate axis labels and scale, as well as label individual lines based on their representative identities. |
JAY VillariUndergraduate student studying Computer Science and Biology at The College of New Jersey Archives
May 2016
Categories |
 RSS Feed
RSS Feed
