Because my program only kept both local and global changes that working towards the desired objective (maximization or minimization), I was instructed to modify it so that it only kept the local pieces with a more desirable ∆G and not to make decisions based on the global ∆G.
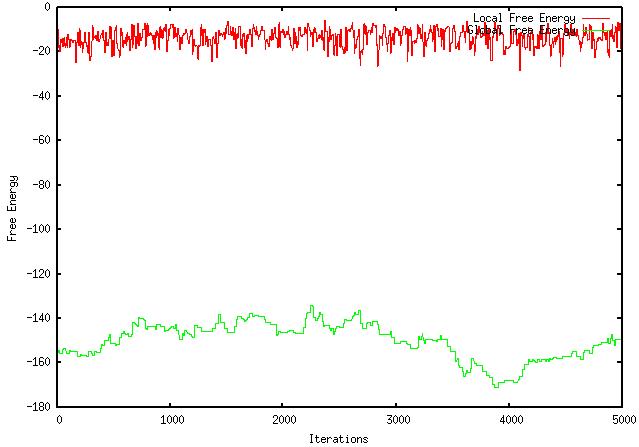
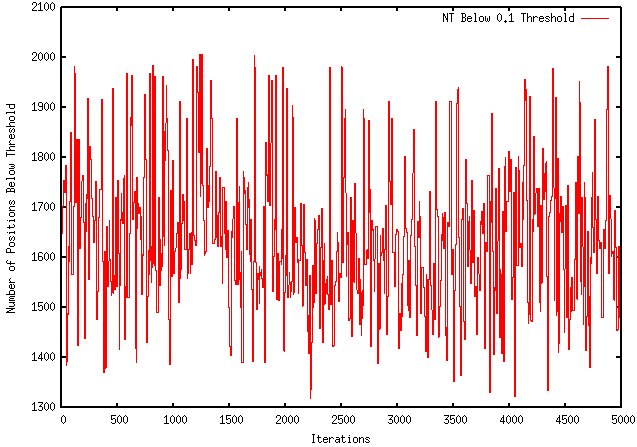
Unfortunately, this modification worsened the extent to which my program optimizes a sequence. For instance, using the GFP protein coding region (whose ∆G = -155.1), my original program maximizes the free energy to -121.4, wheres the modified program only improves it to -149.2. The results are visually represented by the graphs below:
ORIGINAL PROGRAM
Unfortunately, this modification worsened the extent to which my program optimizes a sequence. For instance, using the GFP protein coding region (whose ∆G = -155.1), my original program maximizes the free energy to -121.4, wheres the modified program only improves it to -149.2. The results are visually represented by the graphs below:
ORIGINAL PROGRAM
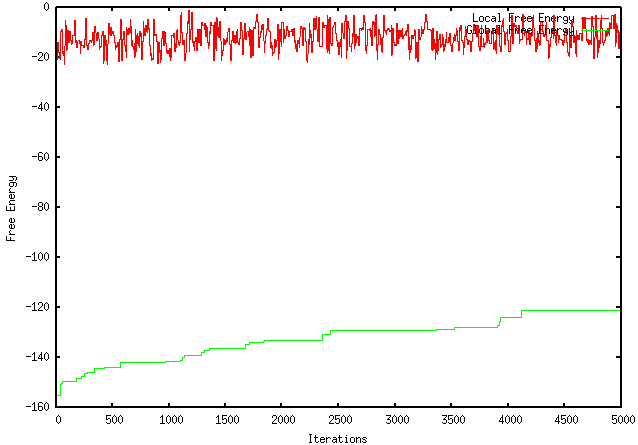
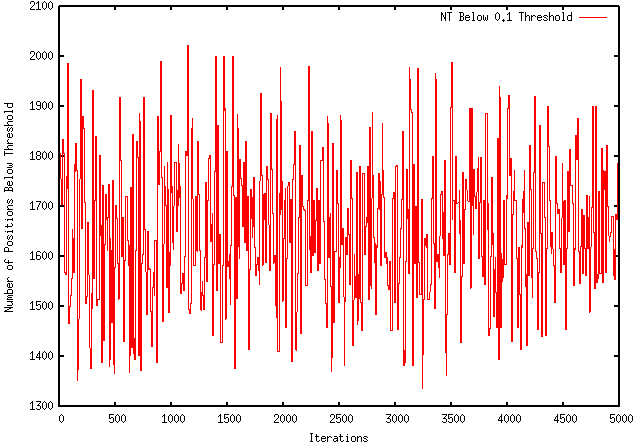
MODIFIED PROGRAM
In addition, when using mRNA Optimiser to optimize GFP, it generates a sequence with a ∆G = -106.8. However, it should be noted, when using mRNA Optimiser to optimize GFP while trying to retain GC-content, it produces a sequence with a ∆G = -126.1. Although my program does not try to retain GC-content, it does have an improved ∆G.
This prompted me to examine mRNA Optimiser's method to get an idea about how they go about maximizing the energy more efficiently. First, a number of codons are selected (in every iteration) and changed for synonymous ones. The ∆G of the new sequence is then calculated and "new sequences with larger values are accepted for the next iteration" (similar to my program). However, there is a significant difference, sequences with lower values are accepted (according to the Boltzmann Distribution) to avoid local maxima. According to the paper, it "allows acceptance of worse solutions at the beginning of the process, slowly decreasing the acceptance probability until only better solutions are chosen as the algorithm approaches the maximum number of iterations". The program ends when 4000 iterations are performed.
To improve the time in which the program runs, a better method for determining MFE was developed (although it produces less accurate results). RNAFold and MFold/UNAFold both have a time complexity of O(n^3) so mRNA Optimiser developed a more simplistic method and improved it to quadratic complexity. The program itself considers all possible single-stem loop conformations and averages their interaction energy. In addition, there is a detailed method to fine-tuning nucleotide interactions. Moreover, two linear regressions are performed to better predict the MFE. The paper can be found at: http://www.ncbi.nlm.nih.gov/pubmed/23325845.
This prompted me to examine mRNA Optimiser's method to get an idea about how they go about maximizing the energy more efficiently. First, a number of codons are selected (in every iteration) and changed for synonymous ones. The ∆G of the new sequence is then calculated and "new sequences with larger values are accepted for the next iteration" (similar to my program). However, there is a significant difference, sequences with lower values are accepted (according to the Boltzmann Distribution) to avoid local maxima. According to the paper, it "allows acceptance of worse solutions at the beginning of the process, slowly decreasing the acceptance probability until only better solutions are chosen as the algorithm approaches the maximum number of iterations". The program ends when 4000 iterations are performed.
To improve the time in which the program runs, a better method for determining MFE was developed (although it produces less accurate results). RNAFold and MFold/UNAFold both have a time complexity of O(n^3) so mRNA Optimiser developed a more simplistic method and improved it to quadratic complexity. The program itself considers all possible single-stem loop conformations and averages their interaction energy. In addition, there is a detailed method to fine-tuning nucleotide interactions. Moreover, two linear regressions are performed to better predict the MFE. The paper can be found at: http://www.ncbi.nlm.nih.gov/pubmed/23325845.
 RSS Feed
RSS Feed
