This week I have made modifications to my program to slightly alter its function. I have made so that, instead of making synonymous changes to all positions above a specified threshold, it only makes a synonymous change to a single position. It then keeps that change, reruns the local sequence in rfold, and makes another single change. Currently, my program iterates 40 times. Below are the results graphed with GNUPlot:
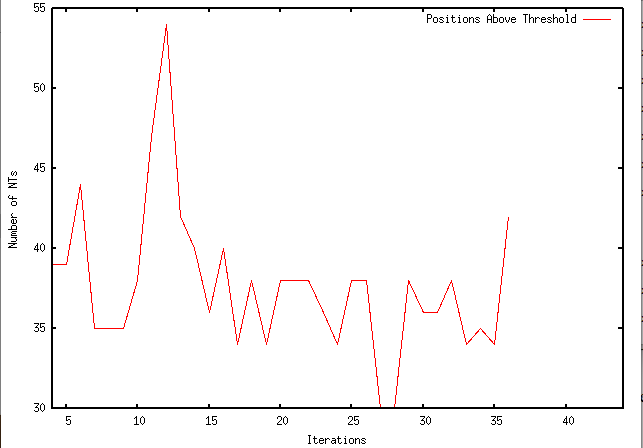
As you can see, the number of nucleotides that exist above the threshold does fluctuate, but generally begins to minimize until the last iteration, in which it suddenly increases to 42 nucleotides above the 0.1 threshold. I aim to experimentally determine the number of iterations that provides the largest decrease in number of nucleotides above the threshold while minimizing the iterations.
I have also gained experience in many other facets of computational research. I learned bash scripting and its applications as well as common ways to configure, install, test and run different (usually biologically related) software. In addition, I have learned more about GNUPlot and how to alter different things. I have figured out how to add x and y axis labels as well as how to add and label a line through all of the data points.
I have also gained experience in many other facets of computational research. I learned bash scripting and its applications as well as common ways to configure, install, test and run different (usually biologically related) software. In addition, I have learned more about GNUPlot and how to alter different things. I have figured out how to add x and y axis labels as well as how to add and label a line through all of the data points.

 RSS Feed
RSS Feed
